Autoimmunerkrankungen sind chronische Entzündungen durch eine Immunreaktion gegen den eigenen Körper. Da Symptome oft unspezifisch und schleichend auftreten, ist die Frühdiagnose meist schwierig.
Autoimmunerkrankungen umfassen eine heterogene Gruppe chronisch-entzündlicher Erkrankungen, deren gemeinsames Kennzeichen eine fehlgeleitete Immunantwort gegen körpereigene Strukturen ist (1,2). Die klinische Präsentation ist häufig unspezifisch, multisystemisch und von schleichendem Beginn, was die frühzeitige Diagnosestellung erschwert.
Auffällig ist jedoch, dass periphere Körperregionen – insbesondere die unteren Extremitäten – nicht selten zu den ersten Orten gehören, an denen sich immunvermittelte Pathologien klinisch manifestieren (4). Veränderungen an Venen, Haut und Weichteilen der Unterschenkel können somit als frühe Indikatoren systemischer Autoimmunprozesse verstanden werden.
Immunologische und vaskuläre Grundlagen
Zentraler pathogenetischer Mechanismus von Autoimmunerkrankungen ist der Verlust der immunologischen Selbsttoleranz. Autoreaktive T- und B-Lymphozyten entgehen regulatorischen Kontrollmechanismen und führen zur Bildung von Autoantikörpern, Immunkomplexen sowie zur chronischen Aktivierung proinflammatorischer Zytokinkaskaden (1). Blutgefäße nehmen hierbei eine besondere Rolle ein. Endothelzellen fungieren nicht nur als physikalische Barriere, sondern sind aktiv an immunologischen Prozessen beteiligt, unter anderem durch die Expression von Adhäsionsmolekülen, Zytokinen und MHC-II-Strukturen (4).
Entzündliche Veränderungen der Gefäßwand werden unter dem Begriff der Vaskulitis zusammengefasst. Diese können primär auftreten oder sekundär im Rahmen systemischer Autoimmunerkrankungen wie dem systemischen Lupus erythematodes oder dem Antiphospholipidsyndrom (3, 12). Die immunvermittelte Endothelschädigung führt zu erhöhter Gefäßpermeabilität, Lumeneinengung, lokalen Ischämien sowie zu einer gesteigerten Thromboseneigung (3).
Warum die unteren Extremitäten besonders betroffen sind
Die besondere Vulnerabilität der Unterschenkel ergibt sich aus einer Kombination anatomischer, hämodynamischer und immunologischer Faktoren. Der venöse Rückstrom aus den Beinen erfolgt gegen die Schwerkraft und ist auf ein intaktes Zusammenspiel von Venenklappen, Muskelpumpe und Gefäßelastizität angewiesen. Bereits geringfügige funktionelle Einschränkungen können zu venöser Stase führen, die in einem entzündlich veränderten Gefäßmilieu die lokale Exposition des Endothels gegenüber Autoantikörpern und Entzündungsmediatoren verstärkt (4,11).
Zudem weist die Haut der Unterschenkel eine hohe Dichte kleiner Gefäße auf, die besonders empfindlich auf immunvermittelte Mikroangiopathien reagieren. Solche Veränderungen werden hier früh klinisch sichtbar, während vergleichbare Prozesse in inneren Organen zunächst asymptomatisch verlaufen können (9,12). Die unteren Extremitäten fungieren damit als peripheres Fenster systemischer Gefäß- und Entzündungserkrankungen.
© designua – adobe.stock.com
Klinische Manifestationen an Beinen und Venen
Autoimmunerkrankungen können sich an den unteren Extremitäten in vielfältiger Weise manifestieren. Besonders häufig betroffen sind Haut, Subkutis und venöse Gefäße. Die klinischen Erscheinungsbilder reichen von funktionellen Durchblutungsstörungen bis hin zu nekrotisierenden Gefäßentzündungen und chronischen Ulzerationen (4,12).
Livedo reticularis und Livedo racemosa
Entzündliche Veränderungen der Gefäßwand werden unter dem Begriff der Vaskulitis zusammengefasst. Diese können primär auftreten oder sekundär im Rahmen systemischer Autoimmunerkrankungen wie dem systemischen Lupus erythematodes oder dem Antiphospholipidsyndrom (3, 12). Die immunvermittelte Endothelschädigung führt zu erhöhter Gefäßpermeabilität, Lumeneinengung, lokalen Ischämien sowie zu einer gesteigerten Thromboseneigung (3).
Die Livedo reticularis tritt gehäuft im Zusammenhang mit dem systemischen Lupus erythematodes, dem Antiphospholipid-Syndrom, Kryoglobulinämien sowie Vaskulitiden kleiner Gefäße auf [6,7,9]. In diesen Kontexten ist sie Ausdruck einer systemischen Gefäßbeteiligung und nicht lediglich ein benignes Hautphänomen.
Die Livedo racemosa stellt eine klinisch bedeutsamere Variante dar. Sie ist durch ein unregelmäßiges, fragmentiertes und großflächigeres Muster gekennzeichnet und persistiert meist unabhängig von äußeren Einflüssen. Sie ist häufiger mit strukturellen Gefäßläsionen, thrombotischen Ereignissen und systemischen Komplikationen assoziiert (9).
Erythema nodosum
Das Erythema nodosum ist die häufigste Form der Pannikulitis und manifestiert sich klinisch durch schmerzhafte, rötliche bis livide Knoten, bevorzugt an den Streckseiten der Unterschenkel (10). Histopathologisch handelt es sich um eine septale Entzündung des subkutanen Fettgewebes ohne primäre Vaskulitis.
Pathogenetisch wird das Erythema nodosum als immunvermittelte Überreaktion auf systemische Entzündungsprozesse interpretiert. Es tritt unter anderem bei Sarkoidose, entzündlichen Darmerkrankungen sowie systemischen Autoimmunerkrankungen auf [10]. Häufig begleiten Allgemeinsymptome wie Fieber, Arthralgien und Müdigkeit das klinische Bild, was den systemischen Charakter widerspiegelt.
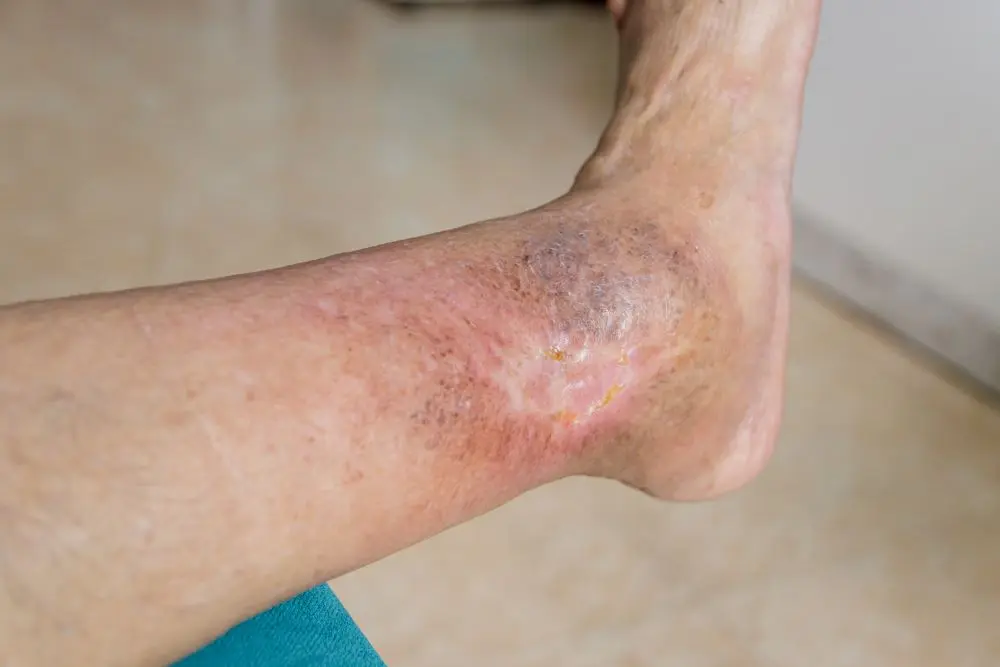
Ulcus cruris im autoimmunologischen Kontext
Neben venösen und arteriellen Ursachen existiert eine relevante Gruppe immunologisch bedingter Ulzera. Diese zeichnen sich durch einen atypischen Verlauf mit schlechter Heilung trotz adäquater Wundtherapie, entzündlich infiltrierten Wundrändern und ausgeprägter Schmerzhaftigkeit aus (11). Sie werden insbesondere bei Vaskulitiden, Kollagenosen und dem systemischen Lupus erythematodes beobachtet (12). Die zugrunde liegende Pathophysiologie umfasst entzündliche Gefäßveränderungen, Mikrothrombosierungen und eine gestörte Geweberegeneration infolge chronischer Ischämie. Die korrekte ätiologische Zuordnung ist essenziell, da diese Ulzera eine systemische immunsuppressive Therapie erfordern (11, 12).
| Klinische Manifestation | Typische Lokalisation | Pathophysiologischer Mechanismus | Assoziierte Autoimmunerkrankungen | Klinische Bedeutung |
| Livedo reticularis | Unterschenkel | Immunvermittelte Gefäßbeteiligung, Mikroangiopathie | SLE, APS, Kryoglobulinämie, kleine Vaskulitiden | Hinweis auf systemische Gefäßbeteiligung |
| Livedo racemosa | Unterschenkel | Strukturelle Gefäßläsionen, Mikrothrombosen | APS, SLE | Erhöhtes Risiko für thrombotische Komplikationen |
| Erythema nodosum | Streckseiten der Unterschenkel | Septale Pannikulitis als immunvermittelte Reaktion | Sarkoidose, CED, systemische Autoimmunerkrankungen | Marker systemischer Entzündungsaktivität |
| Immunologisch bedingtes Ulcus cruris | Knöchelregion, distaler Unterschenkel | Entzündliche Gefäßschädigung, Ischämie, gestörte Regeneration | Vaskulitiden, Kollagenosen, SLE | Hinweis auf systemische Erkrankung, immunsuppressive Therapie erforderlich |
Autoimmunerkrankungen mit besonderer Relevanz für Beinmanifestationen
Systemischer Lupus erythematodes (SLE)
Beim systemischen Lupus erythematodes spielen Immunkomplexablagerungen eine zentrale Rolle. Diese aktivieren das Komplementsystem und führen zu einer entzündlichen Endothelschädigung (6). Klinische Folgen sind Mikrothrombosierungen, Hautvaskulitiden und chronische Durchblutungsstörungen. Die unteren Extremitäten sind besonders häufig betroffen, was sich unter anderem durch Livedo reticularis, Ulzerationen und thrombotische Ereignisse äußert (6, 9).
Antiphospholipid-Syndrom (APS)
Das Antiphospholipidsyndrom ist durch Autoantikörper gegen phospholipidbindende Proteine gekennzeichnet und führt zu einer ausgeprägten Hyperkoagulabilität (7, 8). Klinisch manifestiert es sich durch rezidivierende tiefe Venenthrombosen, Mikrothrombosen der Haut sowie charakteristische Hautveränderungen wie Livedo racemosa. Die unteren Extremitäten sind häufig der Ort der Erstmanifestation (7, 13).
ANCA-assoziierte Vaskulitiden
ANCA-assoziierte Vaskulitiden führen zu nekrotisierenden Entzündungen kleiner Gefäße. Haut- und Beinmanifestationen wie palpable Purpura, Ulzerationen und Nekrosen sind häufig frühe klinische Hinweise auf eine systemische Erkrankung mit potenziell lebensbedrohlichem Verlauf (3,14). Die frühzeitige Erkennung kutaner Zeichen ist daher von hoher diagnostischer Relevanz.
Diagnostische Implikationen
Veränderungen an Beinen und Venen sollten insbesondere bei atypischem Verlauf, fehlendem Therapieansprechen oder rezidivierendem Auftreten Anlass zu einer erweiterten Diagnostik geben. Diese umfasst die Bestimmung von Entzündungsparametern, Autoantikörperprofilen, eine gezielte Gefäßbildgebung sowie gegebenenfalls die histopathologische Untersuchung von Hautbiopsien (4,12). Eine interdisziplinäre Zusammenarbeit zwischen Angiologie, Rheumatologie, Dermatologie und Immunologie ist essenziell.
Fazit
Die unteren Extremitäten sind kein peripherer Nebenschauplatz, sondern ein sensibles Frühwarnsystem systemischer Autoimmunerkrankungen. Veränderungen an Haut und Venen der Beine können entscheidende Hinweise auf immunvermittelte Gefäßprozesse liefern. Ein integrativer diagnostischer Ansatz ermöglicht eine frühere Erkennung und eine gezieltere Therapie mit prognostischem Nutzen für die Betroffenen (4).
Zusammenfassung
Autoimmunerkrankungen entstehen durch den Verlust der immunologischen Selbsttoleranz mit Aktivierung autoreaktiver T- und B-Zellen, Autoantikörpern und chronischer Entzündung. Blutgefäße sind zentral beteiligt, da Endothelzellen immunologisch aktiv sind. Entzündliche Gefäßschäden bis zur Vaskulitis verursachen erhöhte Permeabilität, Lumeneinengung, Ischämien und Thromboseneigung. Die unteren Extremitäten sind besonders anfällig: Der venöse Rückstrom gegen die Schwerkraft begünstigt bei Störungen eine Stase und verstärkt lokale Entzündungen. Mikroangiopathien werden in der gefäßreichen Haut der Unterschenkel früh sichtbar, während innere Organe zunächst symptomarm bleiben. Typische Manifestationen sind Livedo reticularis oder racemosa, Erythema nodosum sowie schmerzhafte, schlecht heilende Ulzera. Sie können auf systemische Erkrankungen wie Lupus erythematodes, Antiphospholipid-Syndrom oder ANCA-assoziierte Vaskulitiden hinweisen. Atypische oder therapierefraktäre Verläufe erfordern eine gezielte weiterführende Diagnostik.
Literatur:
- Rose NR, Mackay IR. The Autoimmune Diseases. 5th ed. London: Academic Press; 2014.
- Fauci AS, Kasper DL, Hauser SL, et al. Harrison’s Principles of Internal Medicine. 21st ed. New York: McGraw-Hill; 2022.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheumatol. 2013;65(1):1–11.
- Merkel PA, Grayson PC. Vascular manifestations of systemic autoimmune diseases. Nat Rev Rheumatol. 2021;17(10):579–594.
- Sunderkötter C, Bonsmann G, Sindrilaru A, et al. Management of leukocytoclastic vasculitis. J Dermatolog Treat. 2018;29(7):1–10.
- Pons-Estel GJ, Ugarte-Gil MF, Alarcón GS. Epidemiology of systemic lupus erythematosus. Expert Rev Clin Immunol. 2017;13(8):799–814.
- Cervera R, Piette JC, Font J, et al. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Medicine (Baltimore). 2002;81(5):355–367.
- Asherson RA. The antiphospholipid syndrome: a review. J Autoimmun. 2000;15(2):113–119.
- Criado PR, Rivitti EA, Sotto MN, et al. Livedo reticularis and livedo racemosa: clinical and pathological review. Clin Dermatol. 2015;33(5):528–536.
- Requena L, Sánchez Yus E. Erythema nodosum. Semin Cutan Med Surg. 2007;26(2):114–125.
- Alavi A, Sibbald RG, Phillips TJ, et al. What’s new: management of venous leg ulcers. J Am Acad Dermatol. 2016;74(4):643–664.
- Sunderkötter C, Rieger S. Cutaneous vasculitis. Rheumatology (Oxford). 2018;57(Suppl 3):iii47–iii55.
- Hughes GRV. Thrombosis, abortion, cerebral disease, and the lupus anticoagulant. BMJ. 1983;287(6399):1088–1089.
- Grayson PC, Ponte C, Suppiah R, et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for ANCA-associated vasculitis. Ann Rheum Dis. 2022;81(3):315–320.
- Wolff K, Johnson RA, Saavedra AP. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 9th ed. New York: McGraw-Hill; 2019
Korrespondenzadresse

© privat
Dr. Christian Moser
Facharzt für Dermatologie, Phlebologie, Proktologie
MVZ Dres. Raulin GmbH, Kaiserstrasse 104, 76133 Karlsruhe
E-Mail: phlebo@mvz-derma-ka.de
Webseite: www.laserklinik.de