Epileptische Anfälle gehören zu den häufigsten neurologischen Begleitsymptomen bei Hirntumoren und stellen sowohl für Betroffene als auch für behandelnde Ärztinnen und Ärzte eine zentrale Herausforderung dar. Dieser Fachartikel bietet einen umfassenden Überblick über die tumorassoziierte Epilepsie (BTRE), deren pathophysiologische Grundlagen, diagnostische Verfahren und moderne Therapieansätze. Mit besonderem Fokus auf praxisrelevante Aspekte richtet sich der Beitrag an Neurologinnen und Neurologen in ambulanten und stationären Versorgungsstrukturen.
Autoren: C. Seliger, C. Schaub
Epidemiologie und klinische Bedeutung
Epileptische Anfälle gehören zu den häufigsten neurologischen Symptomen bei Patientinnen und Patienten mit Hirntumoren. Die Prävalenz variiert dabei erheblich je nach Tumortyp und molekularen Eigenschaften:
Prävalenz nach Tumortyp:
- 30–50 % aller Hirntumorpatientinnen und -patienten entwickeln im Krankheitsverlauf epileptische Anfälle
- Bis zu 80 % bei niedriggradigen Gliomen
- 80–100 % bei long-term epilepsy-associated tumors (LEAT)
Long-term epilepsy-associated tumors (LEAT)
Zu den LEAT zählen unter anderem:
• Gangliogliome
• Dysembryoplastische neuroepitheliale Tumoren (DNET)
• Pilozytische Astrozytome
• Pleomorphe Xanthoastrozytome
• Angiozentrische neuroepitheliale Tumoren
In diesen Entitäten treten epileptische Anfälle in bis zu 80–100 % der Fälle auf und sind häufig das führende klinische Symptom [4, 5].
Niedrigere Anfallsprävalenz bei anderen Tumortypen
Demgegenüber ist die Anfallsprävalenz bei folgenden Tumortypen deutlich geringer und liegt je nach Entität meist zwischen 10 und 50 % [4]:
• Hochgradige Gliome
• Meningeome
• Leptomeningeale Tumoren
• Primäre ZNS-Lymphome
Unterschiede innerhalb der Gliome
Auch innerhalb der Gliome zeigen sich deutliche Unterschiede im Epilepsierisiko:
- Niedriggradige Gliome sind häufiger epileptogen als hochgradige Tumoren
- Oligodendrogliome häufiger als Astrozytome
- IDH-mutierte Gliome häufiger als IDH-Wildtyp-Gliome [6]
Diese Unterschiede unterstreichen die Bedeutung biologischer und molekularer Tumoreigenschaften für die Epileptogenese.
Zeitpunkt des Auftretens
Die Epilepsie kann dabei das Erstsymptom einer bislang unerkannten Neoplasie darstellen oder im Verlauf der Erkrankung, etwa bei Tumorprogression, Rezidiv oder therapieassoziierten Komplikationen, neu auftreten.
Bedeutung für die Lebensqualität
Für die Lebensqualität der Patientinnen und Patienten, aber auch für Prognose und Therapieplanung, spielt die Anfallskontrolle eine zentrale Rolle. Neben der direkten Gefährdung durch Anfälle führen folgende Faktoren häufig zu einem erheblichen Verlust an Autonomie:
- Fahrtauglichkeit
- Berufliche Einschränkungen
- Psychosoziale Belastungen
Versorgungsstrukturen
Für niedergelassene Neurologinnen und Neurologen ist die langfristige Betreuung und medikamentöse Einstellung dieser Patientinnen und Patienten ein wesentlicher Bestandteil der Versorgung, während in klinischen Strukturen insbesondere die Akutdiagnostik, perioperative Betreuung und interdisziplinäre Therapieplanung im Vordergrund stehen.
Der folgende Beitrag gibt einen Überblick über Pathophysiologie, Diagnostik und Therapie der tumorassoziierten Epilepsie (BTRE, brain tumor–related epilepsy) mit besonderem Fokus auf praxisrelevante Aspekte für ambulante und stationäre Versorgungskonzepte.
Pathophysiologie der tumorassoziierten Epilepsie
Die Entstehung epileptischer Anfälle bei Hirntumoren ist multifaktoriell und resultiert aus einem komplexen Zusammenspiel struktureller, neurochemischer, zellulärer und molekularer Veränderungen.
Mechanische und strukturelle Faktoren
Neben der mechanischen Kompression und der infiltrativen Schädigung des Nervengewebes spielen insbesondere Veränderungen im peritumoralen Mikromilieu eine zentrale Rolle für die Epileptogenese.
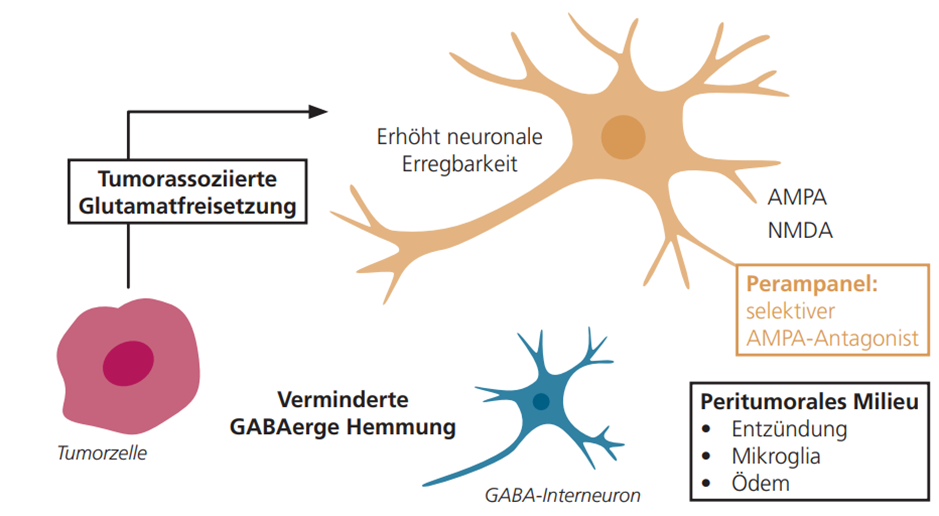
Tumorassoziierte Glutamatfreisetzung
Ein wesentlicher Mechanismus ist die tumorassoziierte Glutamatfreisetzung. Gliomzellen setzen Glutamat in hoher Konzentration frei, was über eine Aktivierung von AMPA- und NMDA-Rezeptoren zu einer gesteigerten neuronalen Erregbarkeit führt.
Parallel ist die inhibitorische GABAerge Transmission im peritumoralen Kortex reduziert, sodass es zu einer pathologischen Verschiebung des Gleichgewichts zwischen Erregung und Hemmung kommt [7, 8].
Weitere beitragende Faktoren
Weitere beitragende Faktoren sind:
- Peritumorale Entzündungsprozesse
- Ischämie
- Ödembildung
- Veränderungen der Ionenkanalexpression
Diese Faktoren senken die kortikale Reizschwelle zusätzlich (▶ Abb. 1) [2].
Molekulare Tumoreigenschaften
Zunehmend wird auch der Einfluss molekularer Tumoreigenschaften auf die Anfallsentstehung erkannt.
IDH1/2-Mutationen
Insbesondere IDH1/2-Mutationen, die vor allem bei diffus infiltrierenden niedriggradigen Gliomen vorkommen, sind mit einer erhöhten Anfallsneigung assoziiert. Der onkometabolische Effekt des Mutationsprodukts D-2-Hydroxyglutarat führt zu einer gesteigerten neuronalen Erregbarkeit und trägt wesentlich zur Epileptogenese bei [9–11].
BRAF-V600E-Mutationen
Auch BRAF-V600E-Mutationen, wie sie insbesondere bei bestimmten glioneuronalen Tumoren auftreten, sind mit einer erhöhten Epilepsieprävalenz verbunden [10, 12].
Klinische Manifestation
Klinisch manifestiert sich Epilepsie besonders häufig bei diffus infiltrierenden, niedriggradigen Gliomen, während sie bei metastatischen oder meningealen Tumoren deutlich seltener auftritt [1, 2].
Bestimmte Hirntumore sind nahezu immer mit einem Anfallsleiden assoziiert, wie z. B.:
- Dysembryoplastischer neuroepithelialer Tumor
- Gangliogliom [4, 5]
Epileptogene Zone
Die epileptogene Zone beschränkt sich meist nicht auf das Tumorgewebe selbst, sondern umfasst häufig auch angrenzende, funktionell noch intakte kortikale Areale, die durch das veränderte Mikromilieu sekundär epileptogen werden.
Klinische Präsentation und Differenzialdiagnose
Epileptische Anfälle bei Hirntumorpatientinnen und -patienten präsentieren sich überwiegend als fokale Anfälle, häufig mit sekundärer Generalisierung.
Generalisierte Anfälle
Generalisierte Anfälle ohne fokalen Beginn sind demgegenüber seltener und sollten immer Anlass zu einer sorgfältigen strukturellen Abklärung geben.
Nicht selten stellen epileptische Anfälle das Erstsymptom eines bislang unbekannten Hirntumors dar, insbesondere bei diffus infiltrierenden Gliomen.
Semiologische Ausprägung nach Tumorlokalisation
Die semiologische Ausprägung der Anfälle hängt maßgeblich von der Tumorlokalisation ab:
Frontallappen
Tumoren im Frontallappen manifestieren sich häufig durch:
- Motorische Anfälle
- Plötzlicher Beginn
- Kurze Dauer
- Komplexe Bewegungsmuster
Parietallappen
Parietale Läsionen führen häufig zu:
- Sensiblen oder sensomotorischen Anfällen
- Parästhesien
- Körperwahrnehmungsstörungen
Temporallappen
Bei temporalen Tumoren dominieren:
- Komplex-fokale Anfälle mit Bewusstseinsstörung
- Automatismen
- Vegetative oder affektive Begleitsymptome
- Déjà-vu-Erlebnisse
- Epigastrische Auren
Okzipitallappen
Okzipitale Läsionen können sich äußern durch:
- Visuelle Phänomene wie Lichtblitze
- Gesichtsfeldausfälle
Differenzialdiagnose
Im klinischen Alltag kommt insbesondere im niedergelassenen Bereich der Differenzialdiagnose eine entscheidende Bedeutung zu.
Nicht-epileptische Ereignisse
Zahlreiche nicht-epileptische Ereignisse können tumorassoziierte Anfälle imitieren:
- Transitorische ischämische Attacken (TIA)
- Synkopen
- Migräne-Auren
- Psychogene nicht-epileptische Anfälle
Anamnestische Hinweise
Eine sorgfältige Anamnese ist dabei oft wegweisend.
Hinweise auf epileptische Genese
Hinweise auf eine epileptische Genese sind unter anderem:
- Typische Aura (was als einfach fokaler Anfall zu verstehen ist)
- Postiktale Phase mit Desorientiertheit oder Müdigkeit
- Lateraler Zungenbiss
- Fremdanamnese, die tonische oder klonische Bewegungen beschreibt
Hinweise auf Synkope
Demgegenüber sprechen folgende Faktoren eher für eine Synkope:
- Rein lageabhängiger Bewusstseinsverlust
- Sehr kurze Bewusstlosigkeit ohne postiktale Phase
- Prodromale vegetative Symptome
Migräne-Auren
Migräne-Auren sind typischerweise:
- Länger andauernd
- Stereotyp
- Häufig von Kopfschmerzen gefolgt
Diagnostische Konsequenzen
Gerade bei erstmalig auftretenden fokalen neurologischen Symptomen ohne klare Zuordnung sollte frühzeitig an eine strukturelle Ursache gedacht und eine zeitnahe bildgebende Diagnostik veranlasst werden.
Eine klare diagnostische Einordnung ist essenziell, da sie nicht nur die epileptologische Therapie, sondern auch die weitere neuroonkologische Behandlung maßgeblich beeinflusst.
Diagnostisches Vorgehen
Die diagnostische Abklärung epileptischer Anfälle bei Verdacht auf eine tumorassoziierte Epilepsie (BTRE) erfordert ein strukturiertes und zeitnahes Vorgehen.
Diagnostische Ziele
Ziel ist es:
- Eine strukturelle Ursache zu identifizieren
- Den epileptischen Fokus einzugrenzen
- Relevante Differenzialdiagnosen auszuschließen
Elektroenzephalogramm (EEG)
Das Elektroenzephalogramm (EEG) zeigt bei BTRE häufig:
- Fokale Verlangsamungen
- Epileptiforme Potenziale
- Irreguläre Aktivität im Bereich des peritumoralen Gewebes
Interpretation der EEG-Befunde
Diese Befunde spiegeln die lokale kortikale Dysfunktion wider, sind jedoch nicht spezifisch und können auch bei anderen strukturellen Läsionen auftreten.
Ein unauffälliges EEG schließt eine tumorassoziierte Epilepsie nicht aus, insbesondere bei:
- Tief gelegenen Tumoren
- Kortikal nicht oberflächlichen Tumoren
Dennoch kann das EEG wertvolle Hinweise auf den epileptogenen Fokus liefern und bei wiederholten oder Langzeitableitungen die diagnostische Sicherheit erhöhen.
Zerebrale Bildgebung
Die zerebrale Bildgebung stellt die entscheidende Säule der Diagnostik dar.
Magnetresonanztomographie (MRT)
Die Methode der Wahl ist die Magnetresonanztomographie (MRT) des Schädels mit Kontrastmittel, da sie die höchste Sensitivität zur Detektion von zerebralen Tumoren besitzt.
Ergänzende Verfahren
Ergänzende Verfahren wie die MR-Spektroskopie können helfen:
- Zwischen neoplastischen und nicht-neoplastischen Läsionen zu differenzieren
- Metabolische Besonderheiten darzustellen
Aminosäure-PET
In ausgewählten Fällen – insbesondere bei unklaren Läsionen oder zur Abgrenzung von Tumorprogress gegenüber therapieassoziierten Veränderungen – kann eine Aminosäure-PET (z. B. FET-PET) zusätzliche diagnostische Information liefern [13, 14].
Vorgehen im ambulanten Setting
Im ambulanten Setting gilt: Nach einem ersten unprovozierten epileptischen Anfall, insbesondere bei fokaler Symptomatik oder persistierenden neurologischen Defiziten, sollte zeitnah eine zerebrale Bildgebung erfolgen.
Überweisung in neuroonkologisches Zentrum
Bei Nachweis oder dringendem Verdacht auf eine strukturelle Läsion ist eine frühzeitige Überweisung in ein neuroonkologisches Zentrum sinnvoll, um eine interdisziplinäre Beurteilung und Therapieplanung zu ermöglichen.
Dies betrifft insbesondere Patientinnen und Patienten mit:
- Neu diagnostizierten Tumoren
- Unklaren Befunden
- Rasch progredienter Symptomatik
Laborchemische Untersuchungen
Ergänzend sind laborchemische Untersuchungen sinnvoll, um metabolische Auslöser oder begleitende Faktoren auszuschließen:
- Elektrolyte
- Entzündungsparameter
Bei bekannter Tumorerkrankung kann die Anfallszunahme zudem ein Hinweis sein auf:
- Tumorprogress
- Ödemzunahme
- Therapieassoziierte Veränderungen
Diese sollten entsprechend weiter abgeklärt werden.
Anfallssupprimierende Therapie bei Hirntumorpatientinnen und -patienten
Die anfallssupprimierende Therapie spielt bei Hirntumorpatientinnen und -patienten eine zentrale Rolle, da das Rezidivrisiko nach einem ersten tumorassoziierten epileptischen Anfall deutlich erhöht ist.
Therapieindikation
Entsprechend wird bei Patientinnen und Patienten mit Hirntumoren bereits nach dem ersten unprovozierten Anfall die Einleitung einer antiepileptischen Behandlung empfohlen.
Wichtig: Eine prophylaktische Gabe ohne Anfallsvorgeschichte wird hingegen nicht empfohlen [14].


