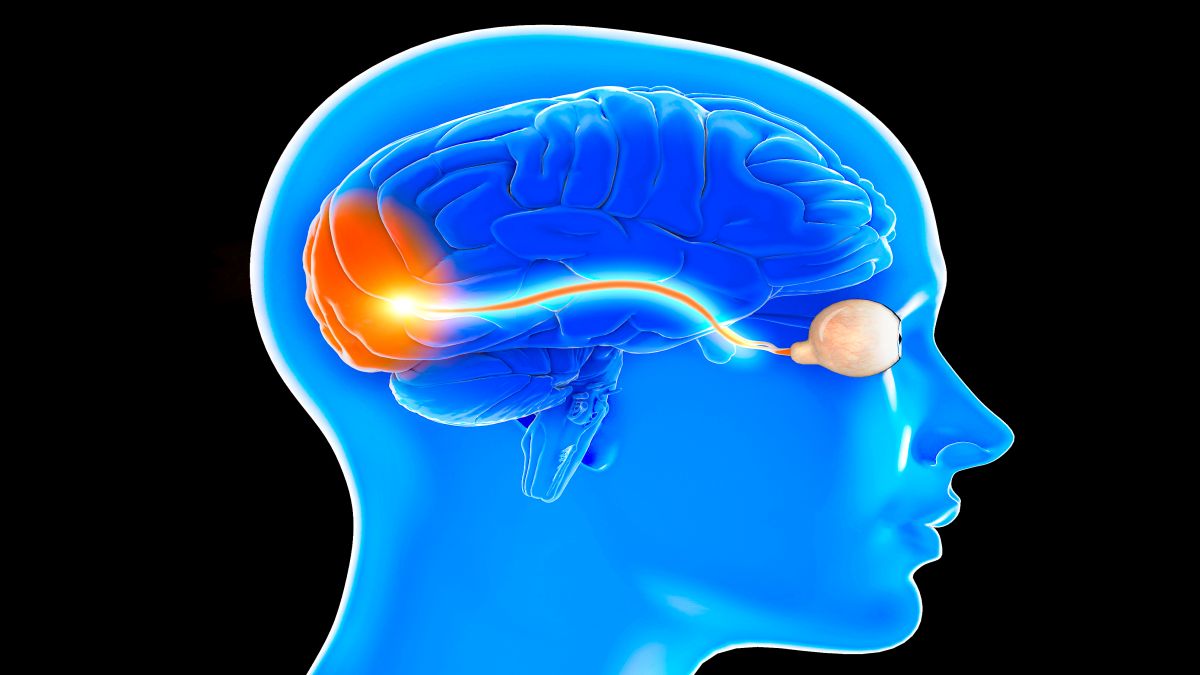
Seit der Erstbeschreibung durch James Parkinson im Jahr 1817 wird die Parkinsonerkrankung (Parkinson’s disease, PD) traditionell als klinisches Syndrom definiert – charakterisiert durch das Vorhandensein typischer Symptome wie Bradykinese, Rigor, Tremor und posturale Instabilität [1]. Über Jahrzehnte galt diese phänotypische Beschreibung, ergänzt durch neuropathologische Befunde post mortem, als Goldstandard der Diagnostik [1]. Die klinischen Diagnosekriterien der Movement Disorder Society (MDS) haben dieses Vorgehen weiter verfeinert, bleiben aber letztlich symptomzentriert [2].
In den letzten Jahren ist jedoch zunehmend deutlich geworden, dass die klinisch definierte PD ein heterogenes Spektrum neurodegenerativer Prozesse umfasst, deren gemeinsamer Nenner nicht immer das klassische neuropathologische Korrelat ist [3]. Diese Diskrepanz zwischen klinischem Erscheinungsbild und zugrunde liegender Biologie hat einen Paradigmenwechsel eingeleitet – weg von der rein klinischen hin zu einer biologisch fundierten Definition der Erkrankung [4].
Treiber dieser Entwicklung sind tiefgreifende Fortschritte in der Biomarkerforschung. Insbesondere die Etablierung der sogenannten Seed Amplification Assays (SAA) hat es erstmals ermöglicht, pathologisch fehlgefaltetes und aggregiertes α‑Synuklein – das Schlüsselmolekül der Lewy‑Pathologie – mit hoher Sensitivität und Spezifität im lebenden Menschen nachzuweisen [5].