Was ist NMOSD?
Die Neuromyelitis-optica-Spektrum-Erkrankung(en) (NMOSD) stehen exemplarisch für ein seltenes, aber klinisch hochrelevantes neuroimmunologisches Krankheitsbild, das in den letzten Jahren einen tiefgreifenden Wandel erlebt hat (zum aktuellen Stand der Diagnostik und Therapie siehe die aktualisierte DGN-Leitlinie zu MS, NMOSD und MOGAD [1]).
Optikusneuritis und Myelitis sind der klinische „Markenkern” der Erkrankung – Symptome, die allerdings auch bei der Multiplen Sklerose (MS) auftreten. Entsprechend wurde die NMOSD um die Jahrtausendwende auch noch vornehmlich als Variante der MS verstanden. Heute wissen wir, dass die NMOSD eine eigenständige primäre Astrozytopathie ist. Die NMOSD ist also keine MS [2].
Die Rolle der Aquaporin-4-Antikörper
Der Nachweis Aquaporin-4-spezifischer Autoantikörper (AQP4-AK) im Serum hat das Verständnis der Pathophysiologie revolutioniert und die diagnostische Präzision erhöht. Werden AQP4-Antikörper nachgewiesen, ist die Diagnose einer NMOSD eindeutig zu stellen („Rule-in”). Die AQP4-AK-negative NMOSD ist dagegen heterogen. Es besteht eine Schnittmenge mit MOG-IgG-assoziierten Erkrankungen (MOGAD), für die Optikusneuritis und Myelitis ebenfalls typisch sind. Bei AQP4-AK-negativen Patientinnen und Patienten mit sicher nachweisbaren MOG-IgG ist daher MOGAD die korrekte Diagnose. Nicht jede rein klinisch definierte NMOSD ist also eine NMOSD im engeren, serologisch definierten Sinn, und die AQP4-AK-negative NMOSD bleibt eine „Rule-out”-Diagnose, bei der weitere pathophysiologische Differenzierungen zu erwarten sind.
Doch trotz aller wissenschaftlichen Fortschritte bleiben grundlegende Versorgungsprobleme bestehen. Diagnostik, Therapie und Versorgungsstrukturen haben sich nicht in gleichem Tempo entwickelt wie das pathophysiologische Verständnis.
Die zentrale Frage lautet daher: Wohin entwickelt sich die NMOSD diagnostisch, therapeutisch und organisatorisch?
Versorgungslandschaft in Deutschland: Eine Black Box
Die NMOSD gilt als seltene Erkrankung („orphan disease”; WHO: Prävalenz < 1/2.000, EU: < 5/10.000).
Überträgt man epidemiologische Daten aus Nachbarländern, ist in Deutschland von wenigen Tausend Betroffenen auszugehen. Die tatsächliche Zahl bleibt jedoch unklar – obwohl die NMOSD im ICD-System (ICD-10: G36.0) seit über 20 Jahren eindeutig kodierbar ist. Hauptgrund ist die fehlende systematische Datenerfassung. Weder Krankenkassen noch Fachgesellschaften bilden die epidemiologische Realität zuverlässig ab. Auch insbesondere als MS verkannte Fehldiagnosen tragen zur Unterschätzung der Prävalenz bei.
Fehlende Transparenz der Versorgungswege
Befragungen zeigen, dass Patientinnen und Patienten mit NMOSD immer noch häufig einen langen Weg bis zur definitiven Diagnose haben. Anfangssymptome wie Optikusneuritis, Myelitis oder Area-postrema-Syndrom werden nicht immer als NMOSD erkannt. Ob Unterschiede in Erfahrung, Ressourcen oder Diagnostik hierfür verantwortlich sind, ist unklar – auch weil nicht systematisch erfasst wird, wer NMOSD-Patientinnen und -Patienten versorgt. Dabei ist eine schnelle Diagnose entscheidend, da jeder Schub bleibende neurologische Schäden verursachen kann.
Uneinheitliche Diagnostik
Die Antikörpertestung ist zentral für die Diagnose. Diese Testung ist aber technisch nicht trivial – nur zellbasierte Assays (CBA) liefern zuverlässige Ergebnisse, sodass auch nur diese als leitliniengerecht gelten.
Wichtige Unterscheidungen bei zellbasierten Assays:
- FCBA (Assays mit fixierten Zellen)
- LCBA (Assays mit lebenden Zellen) – in der Regel höhere Sensitivität
Ob ansonsten nicht mehr empfohlene Testsysteme in der Praxis trotzdem noch Anwendung finden, ist nicht bekannt, aber zu vermuten. Auch über die Vergleichbarkeit der Ergebnisse zwischen Laboren ist wenig bekannt.
Es fehlt also auch bezüglich der Serodiagnostik der NMOSD an Übersicht. Praktisch relevant wird dies, wenn negative Testergebnisse bei fortbestehendem Verdacht laut Leitlinie in spezialisierten Laboren wiederholt werden sollen – die Frage ist aber: wo? Dies gilt umso mehr bei Verdacht auf MOGAD, da hierfür der LCBA als Goldstandard gilt, den nur wenige Speziallabore anbieten.
Fehlende Vernetzung von Behandelnden und Zentren
Die Seltenheit der NMOSD führt dazu, dass viele Neurologinnen und Neurologen kaum je Kontakt mit dieser Erkrankung haben. Erfahrungen mit der Diagnostik, Akut- und Langzeittherapie sind entsprechend ungleich verteilt. Patientinnen und Patienten konzentrieren sich daher in spezialisierten Zentren und nehmen oft lange Wege in Kauf.
Diese Zentren verfügen zwar über die notwendige Expertise, stoßen jedoch bei steigender Nachfrage nach Infusionsprogrammen, Sicherheitsmonitoring und Langzeitnachsorge an Kapazitätsgrenzen. Eine bessere Vernetzung zwischen spezialisierten Zentren und regionaler Versorgung wäre daher notwendig. Strukturmaßnahmen zur Institutionalisierung der Versorgung seltener, ressourcenintensiver neurologischer Erkrankungen wären ein wichtiges strukturpolitisches Ziel.
Neue diagnostische Kriterien: Was ist zu erwarten?
An eine NMOSD sollte insbesondere gedacht werden bei:
- Longitudinal ausgedehnter Myelitis über ≥ 3 Wirbelkörpersegmente (LETM)
- Schwerer ein- oder beidseitiger Optikusneuritis mit deutlichem Visusverlust
- Stammhirnsyndromen mit Bewusstseinsstörungen oder unstillbarem Erbrechen/Schluckauf
- Schlechtem Ansprechen auf eine Glukokortikoid-Pulstherapie
Die 2015 vom International Panel for Neuromyelitis Optica Diagnosis (IPND) publizierten Kriterien bilden derzeit die Grundlage der Diagnosestellung und unterscheiden zwischen AQP4-AK-positiver und -negativer NMOSD [3]. Diese Kriterien werden derzeit zu verschiedenen Aspekten überarbeitet [4], und folgende Anpassungen werden erwartet:
Abgrenzung der MOGAD
Die MOGAD wird heute – trotz klinischer Überschneidungen – eindeutig als eigenständige Erkrankung verstanden und ist mutmaßlich sogar häufiger als die AQP4-AK-positive NMOSD. Frühere „seronegative NMOSD”-Fälle lassen sich nicht selten als MOGAD reklassifizieren [5]. Die neuen Kriterien der NMO werden diese Differenzierung konkret abbilden.
Spezifische MRT-Muster
Zur Frage, welche MRT-Befunde für die NMOSD besonders typisch sind, gibt es seit 2015 neue Erkenntnisse. Die überarbeiteten IPND-Kriterien werden daher weitere MRT-Befunde als typisch für die NMOSD werten:
- T2-hyperintense Läsionen im Hirnstamm im Dach des vierten Ventrikels, im Zwischenhirn um den dritten Ventrikel und im Bereich des Nucleus dentatus des Kleinhirns
- Kontrastmittelaufnahme des Ventrikelependyms
- T2-hyperintense „bright spotty lesions” innerhalb myelitischer Herde
Präzisere biologische Kriterien
Im Zentrum der revidierten Kriterien steht der Antikörperstatus. Im Rahmen der Diagnostik muss daher eine Testung auf AQP4- und ggf. MOG-IgG – vorzugsweise mittels Live-cell-based Assay – erfolgen; ein unbekannter Serostatus ist keine Option mehr.
Bei Nachweis von AQP4-AK fällt das gesamte Spektrum der zu beobachtenden klinischen Manifestationen neu unter den Begriff der Neuromyelitis optica Erkrankung, während doppelt antikörpernegative (AQP4-/MOG-IgG) Manifestationen syndromalen Charakter erhalten (▶ Abb. 1). Die NMOSD wird also eine biologische Diagnose.
GFAP wird vom IPND als vielversprechender (Astrozyten-)Marker angesehen; gleichwohl wird ihm aber (noch) kein Stellenwert bei der Diagnosestellung zugemessen. Das Gleiche gilt für Bestimmung der Neurofilament-Leichtketten (NfL).
Was wird aus der „antikörpernegativen NMOSD”?
Die seronegative NMOSD bleibt diagnostisch herausfordernd, denn diese Gruppe umfasst sowohl Patientinnen und Patienten mit falsch-negativen AQP4-AK-Befunden und MOGAD-Erkrankte, die nicht hinreichend abgeklärt worden sind, als auch korrekt diagnostizierte doppelt-antikörpernegative NMO-Syndrome und klinische Phänokopien.
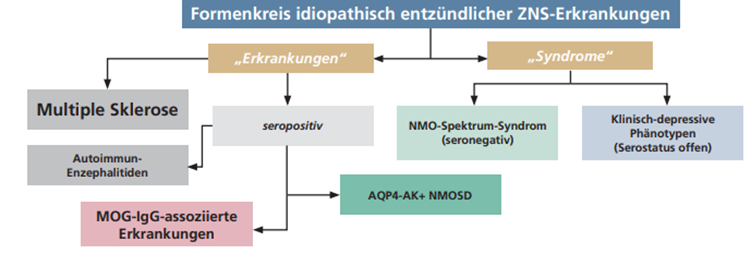
Abb. 1: Die NMOSD in der Landkarte der Neuroimmunologischen Erkrankungen. Revidierte Nomenklatur der NMOSD noch vorläufig.
Bisher wurde postuliert, dass bei tatsächlich doppelt-antikörpernegativen NMO-Fällen weitere, bisher nicht bekannte Autoantikörper gefunden werden. „Klare Treffer” stehen bei dieser Suche aber immer noch aus. Umso mehr werden differenzierte Algorithmen nötig sein, um andere Autoimmunerkrankungen wie etwa das Sjögren-Syndrom, den Systemischen Lupus Erythematodes, die Sarkoidose, paraneoplastische Syndrome oder auch eine optikospinale MS sicher und schnell als Ursache abgrenzen zu können.
Außerdem besteht ein großer therapeutischer Klärungsbedarf. Bisher basiert die Behandlung meist auf klassischen Immunsuppressiva und Rituximab im Off-label-Use. Prospektive Studien unter Einschluss besser definierter doppelt antikörpernegativer Patientinnen und Patienten wären daher dringend erforderlich.